 |
femhrt ® 1/5 is a continuous dosage regimen of a progestin-estrogen combination for oral administration.
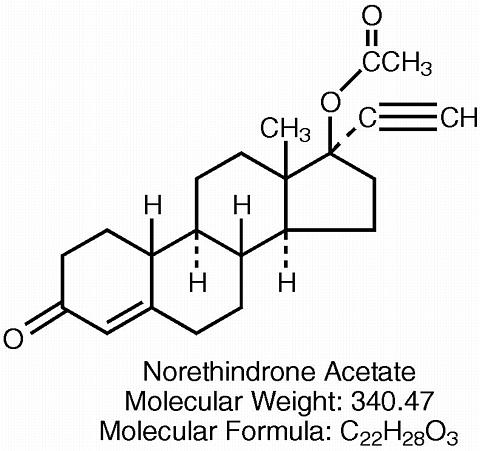
Each white D-shaped tablet contains 1 mg norethindrone acetate [19-Norpregn-4-en-20-yn-3-one, 17-(acetyloxy)-, (17(alpha))-] and 5 mcg ethinyl estradiol [19-Norpregna-1,3, 5(10)-trien-20-yne-3,17-diol, (17(alpha))-]. Each tablet also contains calcium stearate, lactose monohydrate, microcrystalline cellulose, and cornstarch.
The structural formulas are as follows:
|
|
 |
Estrogens are largely responsible for the development and maintenance of the female reproductive system and secondary sex characteristics. Although circulating estrogens exist in a dynamic equilibrium of metabolic interconversions, estradiol is the principal intracellular human estrogen and is substantially more potent than estrone and estriol at the receptor level. The primary source of estrogen in normally cycling adult women is the ovarian follicle, which secretes 70 to 500 mcg of estradiol daily, depending on the phase of the menstrual cycle. After menopause, most endogenous estrogen is produced by conversion of androstenedione, secreted by the adrenal cortex, to estrone by peripheral tissues. Thus, estrone and the sulphate conjugated form, estrone sulphate, are the most abundant circulating estrogens in postmenopausal women. The pharmacologic effects of ethinyl estradiol are similar to those of endogenous estrogens.
Circulating estrogens modulate the pituitary secretion of the gonadotropins, luteinizing hormone (LH) and follicle stimulating hormone (FSH) through a negative feedback mechanism. Estrogen replacement therapy acts to reduce the elevated levels of these hormones seen in postmenopausal women.
Progestin compounds enhance cellular differentiation and generally oppose the actions of estrogens by decreasing estrogen receptor levels, increasing local metabolism of estrogens to less active metabolites, or inducing gene products that blunt cellular responses to estrogen. Progestins exert their effects in target cells by binding to specific progesterone receptors that interact with progesterone response elements in target genes. Progesterone receptors have been identified in the female reproductive tract, breast, pituitary, hypothalamus, bone, skeletal tissue and central nervous system. Progestins produce similar endometrial changes to those of the naturally occurring hormone progesterone.
The use of unopposed estrogen therapy has been associated with an increased risk of endometrial hyperplasia, a possible precursor of endometrial adenocarcinoma. The addition of continuous administration of progestin to an estrogen replacement regimen reduced the incidence of endometrial hyperplasia, and the attendant risk of carcinoma in women with intact uteri.
Norethindrone acetate (NA) is completely and rapidly deacetylated to norethindrone after oral administration, and the disposition of norethindrone acetate is indistinguishable from that of orally administered norethindrone. Norethindrone acetate and ethinyl estradiol (EE) are rapidly absorbed from femhrt 1/5 tablets, with maximum plasma concentrations of norethindrone and ethinyl estradiol generally occurring 1 to 2 hours postdose. Both are subject to first-pass metabolism after oral dosing, resulting in an absolute bioavailability of approximately 64% for norethindrone and 55% for ethinyl estradiol. Bioavailability of femhrt 1/5 tablets is similar to that from solution for norethindrone and slightly less for ethinyl estradiol. Administration of norethindrone acetate/ethinyl estradiol (NA/EE) tablets with a high fat meal decreases rate but not ex-tent of ethinyl estradiol absorption. The extent of norethindrone absorption is increased by 27% following administration of NA/EE tablets with food.
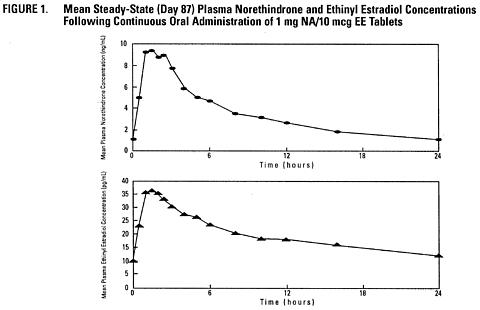
The full pharmacokinetic profile of femhrt 1/5 (1 mg norethindrone acetate/5 mcg ethinyl estradiol) was not characterized due to assay sensitivity limitations. However, the multiple-dose pharmacokinetics were studied at a dose of 1 mg NA/10 mcg EE in 18 post-menopausal women. Mean plasma concentrations are shown below (Figure 1) and pharmacokinetic parameters are found in Table 1. Based on a population pharmacokinetic analysis, mean steady-state concentrations of norethindrone for 1 mg NA/5 mcg EE and 1/10 are slightly more than proportional to dose when compared to 0.5 mg NA/2.5 mcg EE tablets. It can be explained by higher sex hormone binding globulin (SHBG) concentrations. Mean steady-state plasma concentrations of ethinyl estradiol for the 0.5 mg NA/2.5 mcg EE tablets and femhrt 1/5 tablets are proportional to dose, but there is a less than proportional increase in steady-state concentrations for the NA/EE 1/10 tablet.
 |
|
||||||||||||||||||||||||||||||||||||||||||||||||
Based on a population pharmacokinetic analysis, average steady-state concentrations (Css) of norethindrone and ethinyl estradiol for femhrt 1/5 (1 mg NA/5 mcg EE) tablets are estimated to be 2.6 ng/mL and 11.4 pg/mL, respectively.
The pharmacokinetics of ethinyl estradiol and norethindrone acetate were not affected by age, (age range 40-62 years), in the postmenopausal population studied.
Volume of distribution of norethindrone and ethinyl estradiol ranges from 2 to 4 L/kg. Plasma protein binding of both steroids is extensive (>95%); norethindrone binds to both albumin and sex hormone binding globulin (SHBG), whereas ethinyl estradiol binds only to albumin. Although ethinyl estradiol does not bind to SHBG, it induces SHBG synthesis.
Norethindrone undergoes extensive biotransformation, primarily via reduction, followed by sulfate and glucuronide conjugation. The majority of metabolites in the circulation are sulfates, with glucuronides accounting for most of the urinary metabolites. A small amount of norethindrone acetate is metabolically converted to ethinyl estradiol, such that exposure to ethinyl estradiol following administration of 1 mg of norethindrone acetate is equivalent to oral administration of 2.8 mcg ethinyl estradiol. Ethinyl estradiol is also extensively metabolized, both by oxidation and by conjugation with sulfate and glucuronide. Sulfates are the major circulating conjugates of ethinyl estradiol and glucuronides predominate in urine. The primary oxidative metabolite is 2-hydroxy ethinyl estradiol, formed by the CYP3A4 isoform of cytochrome P450. Part of the first-pass metabolism of ethinyl estradiol is believed to occur in gastrointestinal mucosa. Ethinyl estradiol may undergo enterohepatic circulation.
Norethindrone and ethinyl estradiol are excreted in both urine and feces, primarily as metabolites. Plasma clearance values for norethindrone and ethinyl estradiol are similar (approximately 0.4 L/hr/kg). Steady-state elimination half-lives of norethindrone and ethinyl estradiol following administration of 1 mg NA/10 mcg EE tablets are approximately 13 hours and 24 hours, respectively.
femhrt 1/5 is not indicated in children.
The pharmacokinetics of femhrt 1/5 have not been studied in a geriatric population.
The effect of race on the pharmacokinetics of femhrt 1/5 has not been studied.
The effect of renal disease on the disposition of femhrt 1/5 has not been evaluated. In premenopausal women with chronic renal failure undergoing peritoneal dialysis who received multiple doses of an oral contraceptive containing ethinyl estradiol and norethindrone, plasma ethinyl estradiol concentrations were higher and norethindrone concentrations were unchanged compared to concentrations in premenopausal women with normal renal function (see PRECAUTIONS , Fluid Retention ).
The effect of hepatic disease on the disposition of femhrt 1/5 has not been evaluated. However, ethinyl estradiol and norethindrone may be poorly metabolized in patients with impaired liver function (see PRECAUTIONS ).
See PRECAUTIONS , Drug Interactions .
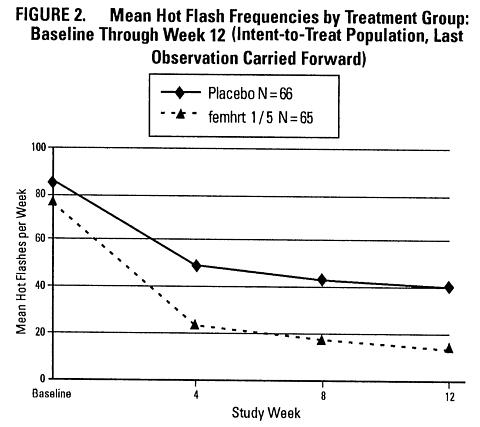
A 12-week placebo-controlled, multicenter, randomized clinical trial was conducted in 266 symptomatic women who had at least 56 moderate to severe hot flashes during the week prior to randomization. On average, patients had 12 hot flashes per day upon study entry.
A total of 65 women were randomized to receive femhrt 1/5 and 66 women were randomized to the placebo group. The efficacy of femhrt 1/5 for the treatment of moderate to severe vasomotor symptoms (VMS) is demonstrated in Figure 2.
 |
A 2-year, placebo-controlled, multicenter, randomized clinical trial was conducted to determine the safety and efficacy of femhrt 1/5 on maintaining bone mineral density, protecting the endometrium, and to determine effects on lipids. A total of 1265 women were enrolled and randomized to either placebo, 0.2 mg NA/1 mcg EE, 0.5 mg NA/2.5 mcg EE, femhrt 1/5 and 1 mg NA/10 mcg EE or matching unopposed EE doses (1, 2.5, 5, or 10 mcg ) for a total of 9 treatment groups. All participants received 1000 mg of calcium supplementation daily. Of the 1265 women randomized to the various treatment arms of this study, 137 were randomized to placebo, 146 to femhrt 1/5, and 141 to EE 5 mcg . Of these, 134 placebo, 143 femhrt 1/5, and 139 EE 5 mcg had a baseline endometrial result. Baseline biopsies were classified as normal (in approximately 95% of subjects), or insufficient tissue (in approximately 5% of subjects). Follow-up biopsies were obtained in approximately 70-80% of patients in each arm after 12 and 24 months of therapy. Results are shown in Table 2.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
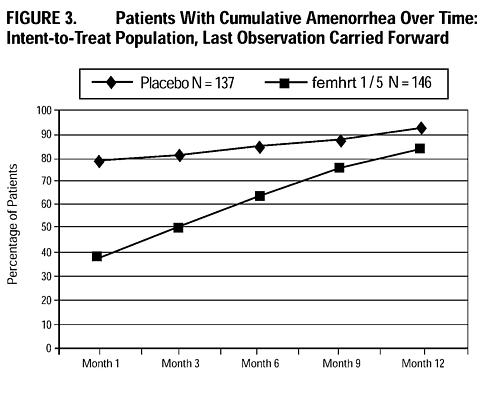
The cumulative incidence of amenorrhea, defined as no bleeding or spotting, was evaluated over 12 months for femhrt 1/5 and placebo arms. Results are shown in Figure 3.
 |
In the 2 year study, trabecular bone mineral density (BMD) was assessed at lumbar spine using quantitative computed tomography. A total of 283 postmenopausal women with intact uteri and normal baseline bone mineral density (124.14 mg/cc ± 9.60 mg/cc) were randomized to femhrt 1/5 (1 mg norethindrone acetate/5 mcg ethinyl estradiol) or placebo, and 87% contributed data to the Intent-To-Treat analysis. All patients received 1000 mg calcium in divided doses. Vitamin D was not supplemented. femhrt 1/5 resulted in significant increases in BMD at each assessment. There was a significant decrease in BMD in the placebo group (see Figure 4).
 |
Patients enrolled in the 2-year osteoporosis and endometrial protection trial were evaluated for changes in lipid parameters after 24 months of therapy. All subjects were postmenopausal women at low risk for cardiovascular disease. Results for femhrt 1/5 and placebo arms are shown in Table 3.
|
|||||||||||||||||||||
femhrt 1/5 is indicated in women with an intact uterus for the:
Since estrogen administration is associated with risks as well as benefits, selection of patients ideally should be based on prospective identification of risk factors for developing osteoporosis. Unfortunately, there is no certain way to identify those women who will develop osteoporotic fractures. Thus, patient selection must be individualized based on the balance of risks and benefits.
Estrogen replacement therapy reduces bone resorption and retards or halts postmenopausal bone loss. Case-control studies have shown an approximately 60% reduction in hip and wrist fractures in women whose estrogen replacement was begun within a few years of menopause. Studies also suggest that estrogen reduces the rate of vertebral fractures. Even when started as late as 6 years after menopause, estrogen may prevent further loss of bone mass for as long as the treatment is continued. When estrogen therapy is discontinued, bone mass declines at a rate comparable to that in the immediate postmenopausal period. There is no evidence that estrogen replacement therapy restores bone mass to premenopausal levels.
Early menopause is one of the strongest predictors for the development of osteoporosis.
The mainstays of prevention and management of postmenopausal osteoporosis are estrogen, an adequate lifetime calcium intake, vitamin D and exercise. Postmenopausal women absorb dietary calcium less efficiently than premenopausal women and require an average of 1500 mg/day of elemental calcium to remain in neutral calcium balance. By comparison, premenopausal women require about 1000 mg/day and the average calcium intake in the USA is 400 to 600 mg/day. Therefore, when not contraindicated, calcium supplementation and adequate daily intake of vitamin D (400 IU) may be helpful.
Progestogens/estrogens should not be used in individuals with any of the following conditions or circumstances:
Based on experience with estrogens and/or progestins:
A causal relationship between estrogen replacement therapy and reduction of cardiovascular disease in postmenopausal women has not been proven. Furthermore, the effect of added progestins on this putative benefit is not yet known.
In recent years many published studies have suggested that there may be a cause-effect relationship between postmenopausal oral estrogen replacement therapy without cyclical progestins and a decrease in cardiovascular disease in women. Although most of the observational studies which assessed this statistical association have reported a 20% to 50% reduction in coronary heart disease risk and associated mortality in estrogen takers, the following should be considered when interpreting these reports:
Occasional blood pressure increases during estrogen replacement therapy have been attributed to idiosyncratic reactions to estrogens. More often, blood pressure has remained the same or has dropped. One study showed that postmenopausal estrogen users have higher blood pressure than nonusers.
Two other studies showed slightly lower blood pressure among estrogen users compared to nonusers. The data on the risk of estrogen use in postmenopausal women and the risk of stroke have not been considered conclusive. Nonetheless, blood pressure should be monitored at regular intervals with estrogen use.
Existing data do not support the use of the combination of progestin and estrogen in postmenopausal women without a uterus.
A complete medical and family history should be taken prior to the initiation of femhrt 1/5 and annually thereafter. These examinations should include special reference to blood pressure, breasts, abdomen, and pelvic organs, and should include a Papanicolaou smear.
Progestin/estrogen therapy may cause some degree of fluid retention. Conditions which might be exacerbated by this factor, such as asthma, epilepsy, migraine, and cardiac or renal dysfunction, require careful observation.
Certain patients may develop undesirable manifestations of estrogenic stimulation, such as abnormal uterine bleeding and mastodynia. In cases of undiagnosed abnormal uterine bleeding, adequate diagnostic measures are indicated (see WARNINGS ).
Estrogens and progestins may be poorly metabolized in patients with impaired liver function. If needed, therapy should be administered with caution.
The pathologist should be advised of progestin/estrogen therapy when relevant specimens are submitted.
Some studies have shown that women taking estrogen replacement therapy have hypercoagulability, primarily related to decreased antithrombin activity. This effect appears dose- and duration-dependent and is less pronounced than that associated with oral contraceptive use. Also, postmenopausal women tend to have changes in coagulation parameters at baseline compared to premenopausal women. There is some suggestion that low dose postmenopausal mestranol may increase the risk of thromboembolism, although the majority of studies (of primarily conjugated estrogen users) report no such increase. There is insufficient information on hypercoagulability in women who have had previous thromboembolic disease, therefore, femhrt 1/5 is contraindicated in such women.
Estrogen therapy may be associated with massive elevations of plasma triglycerides leading to pancreatitis and other complications in patients with familial defects of lipoprotein metabolism.
Patients who have a history of depression should be carefully observed and the drug discontinued if the depression recurs to a serious degree.
Diabetic patients should be carefully observed while receiving progestin/estrogen therapy. The effects of femhrt 1/5 on glucose tolerance have not been studied.
(See Clinical Studies .)
See text of Patient Package Insert which appears after the HOW SUPPLIED section
The following drug/laboratory interactions have been observed with estrogen therapy, and/or femhrt 1/5:
No drug-drug interaction studies have been conducted with femhrt 1/5.
The following section contains information on drug interactions with ethinyl estradiol-containing products (specifically, oral contraceptives) that have been reported in the public literature. It is unknown whether such interactions occur with femhrt 1/5 or drug products containing other types of estrogens.
The metabolism of ethinyl estradiol is increased by rifampin and anticonvulsants such as phenobarbital, phenytoin, and carbamazepine. Coadministration of troglitazone and certain ethinyl-estradiol containing drug products (eg, oral contraceptives containing ethinyl estradiol) reduce the plasma concentrations of ethinyl estradiol by 30 percent.
Ascorbic acid and acetaminophen may increase AUC and/or plasma concentrations of ethinyl estradiol. Coadministration of atorvastatin and certain ethinyl-estradiol containing drug products (eg, oral contraceptives containing ethinyl estradiol) increase AUC values for ethinyl estradiol by 20 percent.
Clinical pharmacokinetic studies have not demonstrated any consistent effect of antibiotics (other than rifampin) on plasma concentrations of synthetic steroids.
Drug products containing ethinyl estradiol may inhibit the metabolism of other compounds. Increased plasma concentrations of cyclosporin, prednisolone, and theophylline have been reported with concomitant administration of certain drugs containing ethinyl estradiol (eg, oral contraceptives containing ethinyl estradiol). In addition, drugs containing ethinyl estradiol may induce the conjugation of other compounds.
Decreased plasma concentrations of acetaminophen and increased clearance of temazapam, salicylic acid, morphine, and clofibric acid have been noted when these drugs were administered with certain ethinyl-estradiol containing drug products (eg, oral contraceptives containing ethinyl estradiol).
Long-term continuous administration of natural and synthetic estrogens in certain animal species increase the frequency of carcinomas of the breast, uterus, cervix, vagina, testis, and liver (see CONTRAINDICATIONS AND WARNINGS ).
Estrogens/progestins should not be used during pregnancy (see CONTRAINDICATIONS AND WARNINGS ).
As a general principle, the administration of any drug to nursing mothers should be done only when clearly necessary since many drugs are excreted in human milk. Estrogen administration to nursing mothers has been shown to decrease the quantity and quality of the milk. Detectable amounts of drug have been identified in the milk of mothers receiving progestational drugs. The effect of this on the nursing infant has not been determined.
Adverse events reported in controlled clinical studies of femhrt 1/5 are shown in Table 4 below.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The following adverse events have been reported with estrogen and/or progestin therapy:
Genitourinary system: changes in vaginal bleeding pattern and abnormal withdrawal bleeding or flow, breakthrough bleeding, spotting, increase in size of uterine leiomyomata, vaginal candidiasis, changes in amount of cervical secretion, pre-menstrual-like syndrome, cystitis-like syndrome.
Breasts: tenderness, enlargement, fibrocystic disease of the breast.
Gastrointestinal: cholestatic jaundice, pancreatitis, flatulence, bloating, abdominal cramps.
Skin chloasma or melasma that may persist when drug is discontinued, erythema multiforme, erythema nodosum, hemorrhagic eruption, loss of scalp hair, hirsutism, itching, skin rash and pruritus.
CNS: headache, migraine, dizziness, chorea, insomnia.
Cardiovascular: changes in blood pressure, cerebrovascular accidents, deep venous thrombosis, and pulmonary embolism.
Eyes: intolerance to contact lenses, sudden partial or complete loss of vision, proptosis, diplopia, otosclerosis.
Miscellaneous: increase or decrease in weight, reduced carbohydrate tolerance, aggravation of porphyria, changes in libido, fatigue, allergic or anaphylactoid reactions, leiomyoma, fibromyoma of the uterus, endometriosis.
Serious ill effects have not been reported following acute ingestion of large doses of progestin/estrogen-containing oral contraceptives by young children. Overdosage of estrogen may cause nausea and vomiting, and withdrawal bleeding may occur.
femhrt 1/5 therapy consists of a single tablet taken once daily.
1. For the Treatment of Vasomotor Symptoms
femhrt 1/5 should be given once daily for the treatment of moderate to severe vasomotor symptoms associated with the menopause. Patients should be reevaluated at 3 to 6 month intervals to determine if treatment is still necessary.
2. Prevention of Osteoporosis
femhrt 1/5 should be given once daily to prevent postmenopausal osteoporosis (see Clinical Studies : Effect on Bone Mineral Density ). Response to therapy can be assessed by measurement of bone mineral density.
Treated patients with an intact uterus should be monitored closely for signs of endometrial cancer, and appropriate diagnostic measures should be taken to rule out malignancy in the event of persistent or recurring vaginal bleeding. Patients should be evaluated at least annually for breast abnormalities and more often if there are any symptoms.
femhrt 1/5 tablets are white and available in the following strength and package sizes:
N 0071-0144-23 Bottle of 90 D-shaped tablets with 1
mg
norethindrone acetate and
5
mcg
ethinyl estradiol
N 0071-0144-45 Blister card of 28 D-shaped tablets with 1
mg
norethindrone acetate
and 5
mcg
ethinyl estradiol
Rx only
Keep this drug and all drugs out of the reach of children.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F)[see USP Controlled Room Temperature].
 |
Your healthcare provider has prescribed femhrt 1/5, a combination of two hormones, a progestin (1 mg norethindrone acetate) and an estrogen (5 mcg ethinyl estradiol) intended for use once a day. This insert describes the major benefits and risks of your treatment, as well as how and when treatment may be taken. If you have any questions, please contact your physician, nurse or pharmacist.
femhrt 1/5 should not be taken in the following situations:
Take your femhrt 1/5 pill once a day at about the same time each day. If you miss a dose, take it as soon as you remember. If it is almost time for your next dose, skip the missed dose and take only your next regularly scheduled dose. Do not take two doses at the same time.
The length of treatment with estrogens varies from woman to woman. You and your healthcare provider should reevaluate every 3 to 6 months whether or not you still need femhrt 1/5 to control your hot flashes.
In addition to the risks and side effects just listed, patients taking estrogen or progestin have reported the following side effects:
If you take femhrt 1/5, you can reduce your risks by carefully monitoring your treatment.
This leaflet provides the most important information about femhrt 1/5. If you want more information, ask your doctor or pharmacist for the professional labeling. The professional labeling is published in a book called "The Physicians' Desk Reference" or PDR, available in bookstores and public libraries.
Revised July 2000
Manufactured by:
DURAMED PHARMACEUTICALS, INC.
CINCINNATI, OH 45213 USA
Distributed by:
PARKE-DAVIS
Division of Warner-Lambert Co. ©1999-'00
Morris Plains, NJ 07950 USA
0132G032